 HOME
HOME
 Research Top
Research Top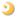 E. coli auxotrophic expression strains
E. coli auxotrophic expression strains 古細菌 Archaea | Sulfurisphaera tokodaii (f. Sulfolobus tokodaii)
古細菌 Archaea | Sulfurisphaera tokodaii (f. Sulfolobus tokodaii) モデル鉄硫黄蛋白質|Fe-S world in Sulfolobus
モデル鉄硫黄蛋白質|Fe-S world in Sulfolobus ISC-like ferredoxin (FdxB) from P. putida
ISC-like ferredoxin (FdxB) from P. putida Microbial "mitoNEET" homologs
Microbial "mitoNEET" homologs Archaeal respiratory complex II
Archaeal respiratory complex II 複合金属酵素の発現
複合金属酵素の発現 Ligand mutagenesis
Ligand mutagenesis 鉄硫黄酵素の分光学
鉄硫黄酵素の分光学 金属コファクターの分子進化
金属コファクターの分子進化 ICC Project
ICC Project
日本医科大学、医学部、生化学・分子生物学(代謝・栄養学)所属の 岩崎 俊雄グループでは、好熱菌等を主材料とした各種金属酵素の構造機能進化や、アミノ酸要求性の新規大腸菌発現宿主株作成につき、国内外の様々な研究機関と共同研究を実施しています。
 Publication List
Publication List Go to the Research Top Page
Go to the Research Top Page"Iron-sulfur (Fe-S) world" in aerobic and thermoacidophilic archaea Sulfolobus
The structure of a metal site in metalloenzymes critically influences the fine-tuning of redox and/or catalytic activities in biology. The substitution and/or displacement events at the local metal-binding site(s) in a protein might have greatly enhanced their capabilities of conducting a wide range of unique redox chemistry in biological electron transfer conduits which often use a limited number of basic protein scaffold. Iron-sulfur (Fe-S) cluster prosthetic groups, consisting of nonheme iron and acid-labile inorganic sulfide atoms, are functionally highly versatile and may be among the most ancient modular metallo-cofactors. These cofactors sustain biologically and evolutionary indispensable processes in the early days of primitive life on earth, such as electron transfer, substrate binding/activation in the hydrogen, nitrogen, carbon and sulfur metabolisms, anaerobic respiration, and photosynthesis - some of the most complicated reactions in the chemistry of life processes. Among protein-bound Fe-S redox sites, which usually contain terminal sulfur ligands from cysteinyl groups, the mononuclear Fe atom in a tetrahedral environment of S ligands is the simplest form, as seen in the rubredoxin family. Other major forms are polynuclear clusters, such as those containing [2Fe-2S], [3Fe-4S], [4Fe-4S], or [8Fe-7S] core units, found in a variety of ferredoxins and complex Fe-S enzymes. In addition to their electron transfer roles, Fe-S proteins are also known to participate in substrate binding/activation, environmental sensing and gene regulation, and more recently are suggested to be potentially involved in several human diseases (e.g. Parkinson's disease, Friedreich's ataxia). The general importance of these Fe-S cofactors in biology is largely attributable to specific features of iron and sulfur chemistry. Therefore the assembly and interplay of the Fe-S cluster core with the surrounding protein is the key to in-depth understanding of the underlying mechanisms.
The archaeal domain contains organisms having the most extraordinary optimal growth conditions, with members flourishing at the extremes of pH, temperature, and salinity. The majority of thermophilic archaea are anaerobic organisms, because oxygen is often scarce in their habitats. Biochemical and genetic evidence indicates that one of the characteristic features in the central metabolic pathways of both anaerobic and more unusual aerobic archaea is the involvement of small modular Fe-S proteins called ferredoxins in electron transport, and many Fe-S enzymes are produced as well in the cells. The cytoplasm of chemoheterotrophically-grown aerobic and thermoacidophilic archaea such as Sulfolobus tokodaii and Thermoplasma acidophilum is in fact remarkably enriched with bacterial-type ferredoxin(s), containing at least ~10 times more than some aerobic and thermophilic "fast-clock" bacteria such as Thermus thermophilus HB8 (unpublished results).
The variation of a common theme in the ferredoxin-dependent pathways of anaerobic and aerobic archaea is striking; especially considering the early days of living organisms on earth, during which the atmosphere became progressively oxidative due to the emergence of photosynthesis by cyanobacterial ancestors that lead to the prevalence of environmental iron in the trivalent state. Under these conditions microaerobic archaeal ancestors had to adapt to the circumstances where, in some cases, the concentration of soluble iron compounds is below their physiological demands. Diminishing iron levels posed a serious challenge for early aerobic archaea. Additionally, the polynuclear Fe-S cluster prosthetic groups contain "acid-labile" sulfide bridges, which are inherently unstable at very acidic conditions. The stunning capability of some contemporary aerobic and thermoacidophilic archaea to grow at extremely low pH with intact Fe-S clusters has implicit meaning in that the intracellular Fe-S world must be protected not only by scavenging reactive oxygen species, but also by balancing its intracellular pH at an acceptable value in the face of a huge proton gradient.
up
Archaeal zinc-containing ferredoxins and related enzymes
The physiological significance of bacterial-type ferredoxins in the aerobic and thermoacidophilic archaea, such as Sulfolobus and Thermoplasma, was first recognized by Kerscher et al., who demonstrated that ferredoxins are abundant in the cytoplasm and serve as an effective electron acceptor of a coenzyme A-acylating 2-oxoacid:ferredoxin oxidoreductase. It is a key Fe-S enzyme of the oxidative tricarboxylic acid cycle and of coenzyme A-dependent pyruvate oxidation in aerobic archaea. This oxidation takes the place in a NAD+-dependent 2-oxoacid dehydrogenase multienzyme complex found in most aerobic and mesophilic bacteria and eukarya. Many 2-oxoacid:ferredoxin oxidoreductase paralogs have been identified in hyperthermophilic archaea and bacteria, some of which participate in the ferredoxin-dependent peptide fermentation pathways. It remains to be established how enzymatically reduced ferredoxin is reoxidized in aerobic and thermoacidophilic archaea.
The X-ray crystal structure of the A2-type pyruvate:ferredoxin oxidoreductase from Desulfovibrio africanus has been determined and shown to contain one thiamine pyrophosphate, one Mg2+, and three [4Fe-4S] clusters as prosthetic groups per protomer. The ab-/a2b2-type homologs from aerobic archaea inherently lack the Fe-S subunit/domain called δ, which harbors two [4Fe-4S] clusters, presumably as an evolutionary consequence of one protein adaptation strategy occurring under permanently oxidative conditions. Likewise, the superfamily of archaeal and bacterial 2-oxoacid:ferredoxin oxidoreductases have different molecular sizes and subunit compositions and exhibit highly mosaic structures with respect to their domain/subunit arrangements. This implies that they might have evolved through multiple gene duplication, fusion, and reorganization events of primordial smaller fragments. Notably, many other Fe-S enzyme complexes in biology seem to have evolved by modular evolution in an analogous way, through which the representative superfamily has become functionally divergent to meet the physiological demands.
Major ferredoxins from chemoheterotrophically-grown aerobic and thermoacidophilic archaea such as Sulfolobus and Thermoplasma, which serve as electron acceptors of 2-oxoacid:ferredoxin oxidoreductases, are characterized by relatively higher molecular masses for bacterial-type ferredoxins (~12-16 kDa) because of a long N-terminal extension region and central loop attached to the ferredoxin core fold. Unlike small [4Fe-4S] ferredoxins from some anaerobic and hyperthermophilic archaea such as Pyrococcus furiosus and Thermococcus profundus, they harbor one each of low-potential [3Fe-4S]1+,0 and [4Fe-4S]2+,1+ clusters (Fig. 1). The most unusual feature of these ferredoxins is the presence of an isolated zinc center, and hence they are called the "zinc-containing ferredoxins" (Fig. 2).
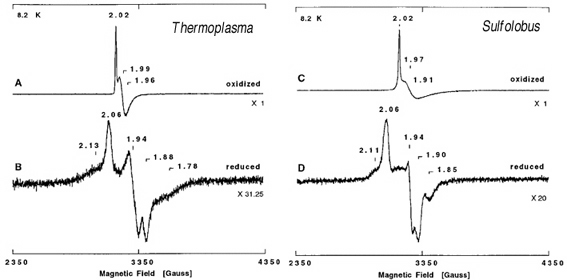
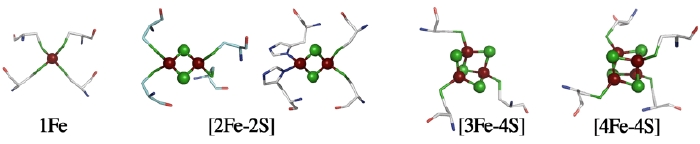
Figure 1. X-band EPR spectra of zinc-containing ferredoxin from T. acidophilum strain HO-62 (a and b) and S. tokodaii strain 7 (c and d) in the air-oxidized (a and c) and dithionite-reduced (b and d) states at pH=9.3. Upon reduction of these ferredoxins by excess dithionite under anaerobic conditions, the sharp g=2.02 EPR signals (a, c) disappeared, and a broad low field resonance at g~12 appeared; this signal is characteristic of the reduced S=2 [3Fe-4S]0 cluster (data not shown). In addition, rhombic EPR signals at g=2.06, 1.94, and 1.88 (b) and g=2.06, 1.94, and 1.90 (d), both attributed to a reduced S=1/2 [4Fe-4S]1+ cluster, were detected up to 30 K for T. acidophilum and S. tokodaii ferredoxins, respectively, together with additional wings on the high and low field sides of the main EPR signals due to magnetic interactions with the reduced S=2 [3Fe-4S]0 cluster. For details, see the original article of this work.
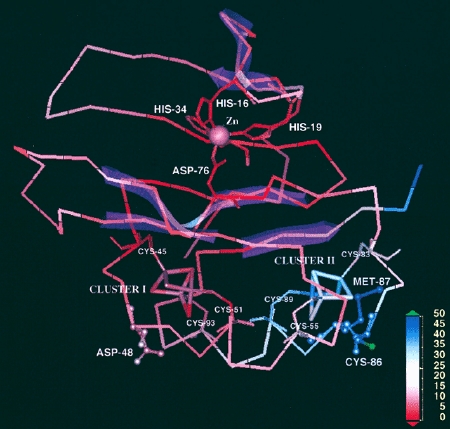
Figure 2. The 2.00-Å resolution crystal structure of the 6Fe-containing form of zinc-containing ferredoxin from Sulfolobus tokodaii strain 7 (PDB code 1XER.pdb), colored by the temperature factors. The metal centers (zinc, cluster I, and cluster II) and important residues are labeled, and beta-sheet structures are shown transparently in pink. In this structure, the region with the highest average temperature factors (blue) is in the vicinity of the [3Fe-4S] cluster II, especially around Cys-86 and Met-87, whereas other part mostly shows lower temperature factors (red). This figure was prepared using Insight II (Molecular Simulations Inc.).
The isolated zinc site was first identified by the 2.00-Å structure of the S. tokodaii ferredoxin (PDB code, 1xer.pdb) in conjunction with the metal content analysis done by our group. It is tetrahedrally coordinated with three histidine imidazole groups (contributed by His16, His19, and His34 in the N-terminal extension region, consisting of three β-strands and one α-helix) and one carboxylate group (contributed by Asp76 in the ferredoxin core fold). The isolated zinc site is buried within the molecule (about 5 Å deep from the protein surface), in the boundary between the N-terminal extension and the cluster-binding ferredoxin core fold, connecting these together. Subsequently, the zinc K-edge X-ray absorption spectroscopic analysis has shown the presence of an isolated and structurally conserved zinc center in S. tokodaii and T. acidphilum zinc-containing ferredoxins (Fig. 3). This center is tetrahedrally coordinated with (most likely) three histidine imidazoles and one carboxylate, with the average Zn-N bond distance of 2.01 Å and the Zn-O bond distance in the range 1.89-1.94 Å. These values are very similar to the average crystallographic Zn-N and Zn-O bond distance of 1.96 Å and 1.90 Å, respectively, in the S. tokodaii zinc-containing ferredoxin structure (PDB code, 1xer.pdb). The sequence comparisons suggest that three histidine residues in the N-terminal extension region and one conserved aspartate in the ferredoxin core fold are strictly conserved in all archaeal zinc-containing ferredoxins, which suggests that they probably serve as ligands to the isolated zinc center. Although the isolated zinc site apparently contributes in part to ferredoxin thermal stability, zinc-lacking isoforms, e.g. from Sulfolobus metallicus and Acidianus ambivalens, have devised a natural strategy that accounts for an enhanced thermal stability without using the zinc site.
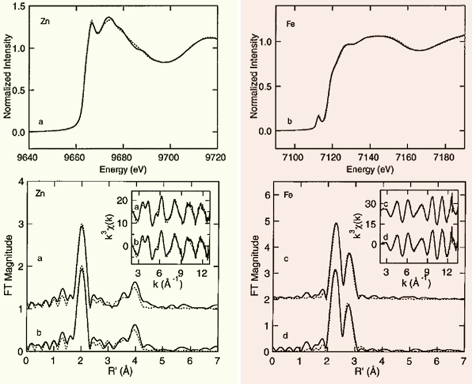
Figure 3. (Top) Zinc (left) and iron (right) x-ray absorption spectra of zinc-containing ferredoxin from T. acidophilum (solid line) and S. tokodaii (dotted line). (Bottom left) k^3-weighted zinc EXAFS (inset) and Fourier transforms of T. acidophilum (a) and S. tokodaii (b) zinc-containing ferredoxins (solid line) and predicted results for Zn(imid)4(COO-) (dashed line). (Bottom right) k^3-weighted iron EXAFS (inset) and Fourier transforms of T. acidophilum (a) and S. tokodaii (b) zinc-containing ferredoxins (solid line) and predicted results for FeS4Fe2 (dashed line). This figure was prepared by Dr. Nate Cosper, Dr. Christina Stålhandske, and Prof. Robert Scott (University of Georgia). Curve-fitting analyses are given in the original article of this work. The XAS data were collected at SSRL, which is operated by the US DOE, Division of Chemical Sciences (the SSRL Biotechnology program is supported by the NIH, Biomedical Resource Technology Program, Division of Research Resources).
The overall protein fold of archaeal zinc-containing ferredoxins is largely asymmetric due to the presence of a long N-terminal extension and the insertion of central loop region (Fig. 2). Nevertheless, the ferredoxin core fold shows the strict conservation of a pseudo-two-fold symmetry with respect to the local two Fe-S cluster binding sites, as typically found for the bacterial-type 8Fe-containing dicluster ferredoxins harboring two [4Fe-4S]2+,1+ clusters. It seems reasonable to postulate that early zinc-containing ferredoxins might have evolved as an 8Fe-containing two-electron carrier without zinc, to which the N-terminal extension and central loop regions were attached in the later stage of modular evolution. Interestingly, zinc-containing ferredoxins exhibit limited distribution in the archaeal domain (such as the Sulfolobales, Halobacteriales, and Thermoplasmatales) at the genomic sequence level, and up until now have been purified exclusively from the aerobic and thermoacidophilic archaea such as Sulfolobus and Thermoplasma. It is emphasized that, in thermophilic euryarchaeotes, zinc-containing ferredoxin has been isolated as a major ferredoxin from the Thermoplasmatales but not Halobacteriales, an unexpected result based on the universal 16S rRNA-based phylogenetic tree. Analogous observation has been reported for the functionally equivalent ferredoxins of extremely halophilic and aerobic euryarchaeotes, which contain a single plant-type [2Fe-2S] cluster and exhibit the sequence similarity to those of extremely halophilic cyanobacteria. It should be noted that the bacterial-type and (usually more oxygen-tolerant) plant-type ferredoxins are totally unrelated at the primary and tertiary structural levels. These observations lend credence for possible phylogenetic distribution of these archaeal ferredoxin genes driven in part by horizontal (lateral) gene transfer in the extreme environments.
up
Intracellular pH matters
Contemporary aerobic and anaerobic archaea apparently inherited the intracellular Fe-S world from their anaerobic ancestors, which can be explained by the emergence of Fe-S clusters as central catalysts of metabolism from when life had evolved in an anaerobic environment. The stunning capability of some aerobic and thermoacidophilic archaea to grow at extremely low pH has therefore implicit meaning in that the intracellular Fe-S world must be protected not only by scavenging reactive oxygen species, but also by balancing the intracellular pH at an acceptable value (typically 5.6-6.5 in Sulfolobus and Thermoplasma) in the face of a huge proton gradient (ΔpH = pHin - pHout). The ΔpH across the cytoplasmic membrane of these archaea is intrinsically linked to the cellular energetics, because it is the primary contributor to the proton motive force (PMF):
PMF = ΔΨ - 2.3(RT/F) ΔpH (mV)
where ΔΨ is the membrane potential generated by the transport of electrical charge, R the gas constant, T the absolute temperature (K), and F the Faraday constant (the effect of 1 unit pH difference is 2.3(RT/F), which equals 59 mV at 25 °C and 70 mV at 80 °C). However, the influx of protons through the archaeal AoA1-ATP synthase upon ATP production intensifies cellular protonation, and therefore need to be balanced by extrusion using the cognate proton translocating systems to remove excess protons from the cytoplasm [otherwise, this would simply result in rapid acidification of the cytoplasm and dissipate any ΔpH formation across the membrane]. Thus, the balance between the proton permeability across the membrane (kept very low in thermoacidoacidophiles), the proton influx through the energetic and transport systems, and the rate of outward proton pumping determines whether an archaeal cell can sustain an appropriate PMF. The mechanistic detail of how this thin-line balance could be achieved has not been clearly understood.
A mechanism used by some acidophilic archaea such as Thermoplasma to reduce the proton influx is the generation of an inside positive ΔΨ, which is opposite to the inside negative ΔΨ in mammalian mitochondria. It is suggested that the reversed ΔΨ is generated by a difference in electrical potential (Donnan potential) formed between a greater influx of cations (such as potassium ions) and the outward flux of protons. This is in line with our preliminary study on the aerobic respiratory chain of T. acidophilum, which contains at least cytochrome bd as a respiratory terminal oxidase (unpublished results) that is usually not a proton pump. However, this concept is apparently not applicable to Sulfolobus, where the inside negative ΔΨ is rather low (about -20 to -40 mV at 45 °C) and the PMF is largely composed of a ΔpH of greater than 2 units.
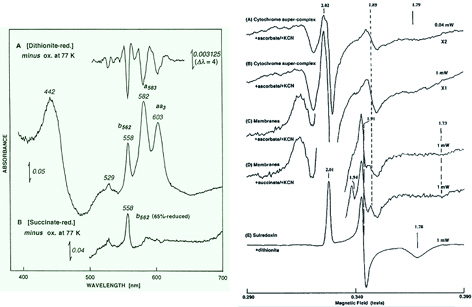
Figure 4. (Left panel) The low temperature optical spectra of the Sulfolobus tokodaii membranes. (A) Dithionite-reduced minus oxidized difference spectrum at 77 K of the archaeal membrane-bound a- and b-type cytochromes in 60 mM potassium phosphate buffer, pH 6.8. No c- and d-type cytochromes were detected. The low temperature second-order finite derivative spectrum (top) of the archaeal membranes suggests the absence of any split α-band in the b-type cytochrome region. (B) 5 mM succinate-reduced minus oxidized difference spectrum at 77 K of the archaeal membrane-bound cytochromes. Upon addition of 5 mM succinate to the S. tokodaii membranes at room temperature in the absence of any respiratory inhibitor, cytochrome b562 was partially reduced (~65% in the case of this figure) while the bulk of the a-type cytochromes remained in the oxidized state. (Right panel) EPR spectra at 10 K of the purified cytochrome supercomplex reduced by ascorbate in the presence of 2 mM cyanide (A and B) and the S. tokodaii membranes reduced by sodium ascorbate (C) and succinate in the presence of 5 mM cyanide (D). The g values and the relative intensity scales are indicated in the figure. In the cyanide-treated reduced membranes, the additional gy=1.91 Rieske-type [2Fe-2S] center could be detected (C and D). The EPR spectrum of the dithionite-reduced form of the cognate “sulredoxin” (a water-soluble, high-potential Rieske [2Fe–2S] protein from S. tokodaii) is shown for comparison (E). These figures were adapted from the original article of this work.
The Sulfolobus species have the unusual terminal oxidase segments of the aerobic respiratory chain, consisting mainly of only a- and b-type cytochromes (Fig. 4), which most likely fulfill the role as an effective proton pump in vivo and preserve the cognate Fe-S world descendant from their anaerobic ancestors. Primary dehydrogenases, some of which are complex Fe-S enzymes, provide the reducing equivalents (from the respiratory substrates such as succinate, NADH, and sulfide) to the central caldariellaquinone pool in the tetraetherlipid monolayer membrane. It should be commented here, however, that most of key biochemical/genetic characterization of the Sulfolobus respiratory chains were carried out before the availability of a variety of the genome-wide sequence information and the mechanistically insightful 3D structures of the terminal segments of the tractable respiratory complexes from bacteria and eukaryal mitochondria. In retrospect, many experimental data in the literature from the pre-genomic era were discussed in seemingly oversimplified ways, perhaps inspired by an idea that an archaeal aerobic respiratory chain might be "primitive and simple". The archaeal respiratory chains may be in fact archaic, but not so primitive as they had seemed two decades ago. For instance, while the terminal oxidase supercomplexes of Sulfolobus acidocaldarius (SoxABCD and SoxM supercomplexes) and S. tokodaii have been shown to mimic a genetic and functional fusion of mitochondrial respiratory complexes III and IV, the number of the redox centers resolved spectroscopically in the literature is insufficient to explain the proposed intramolecular electron transfer mechanism, particularly in the light of a modified Q-cycle scheme of respiratory complex III, which is characterized by bifurcation of electron transfer between two different acceptor chains that allows coupling to proton transfer. Thus, the Sulfolobus aerobic respiratory chain in a mechanistic context is still in its infancy compared with the mitochondrial and bacterial tractable model systems, and needs to be explored in future studies.
up
Formation of intracellular Fe-S clusters does not occur spontaneously but requires specific biosynthetic pathways
In contemporary bacteria and eukarya, the de novo Fe-S cluster biogenesis and maturation in vivo have been shown to require specific enzymes in the carefully regulated Fe-S cluster biosynthesis systems, while spontaneous assembly of the Fe-S clusters does occur in vitro. At least three types of the Fe-S cluster biosynthesis systems (ISC (iron sulfur cluster), SUF (mobilization of sulfur), and NIF (nitrogen fixation)) are known, with significant variations in terms of the phylogenetic distribution. For example, in Escherichia coli the ISC pathway is the major system for in vivo Fe-S cluster assembly, compared to the SUF pathway. In the eukaryal domain, ISC homologs are found to be localized largely in mitochondria, while SUF homologs are found in some chloroplasts. It is therefore possible to postulate that the mitochondrial ISC system originated from the endosymbiotic bacterial ancestor and the plastid SUF system from the cyanobacterial ancestor. In these tractable model organisms, the regulation of biological Fe-S cluster assembly is further complicated by the involvement of other accessory proteins required for the in vivo function, and is not fully understood.
The common concept of the three de novo Fe-S cluster biosynthesis systems is that in vivo cluster assembly requires at least (i) cysteine desulfurases (such as NifS, IscS and SufS) and (ii) Fe-S cluster scaffold proteins (such as IscU, IscA, SufU, and likely SufBCD) with the capacity to construct transient [2Fe-2S] or [4Fe-4S] clusters and then transfer Fe-S clusters to target apo-proteins (as schematically illustrated in Fig. 5). While pyridoxal phosphate-containing cysteine desulfurases utilize L-cysteine for mobilization of S for Fe-S core formation, there is as yet no consensus concerning immediate iron donor for Fe-S cluster assembly. The ISC machinery has been most closely investigated, and bacterial genomic sequence analyses showed the relatively conserved gene arrangement as iscR-iscS-iscU-iscA-hscB-hscA-fdx (IscR is a transcriptional regulatory protein, HscA/HscB DnaK/J-type heat-shock proteins, and Fdx an adrenodoxin-like ferredoxin).
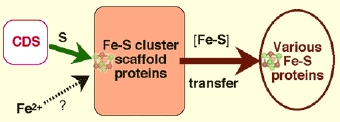
Figure 5. Schematic illustration of the cysteine desulfurase (CDS)-mediated, transient Fe-S cluster assembly on Fe-S cluster scaffold proteins and subsequent cluster transfer to various target apoproteins.
The importance of the SUF machinery in the Fe-S cluster biosynthesis function was clarified in E. coli by construction of different combinations for altered expression of the isc and suf operons. Disruption of the E. coli suf operon does not cause any major defects, whereas the loss of both the ISC and SUF systems leads to synthetic lethality. The components of the suf operon has been shown to be preferred for Fe-S cluster biosynthesis under oxidative stress conditions and during iron starvation; although, the ISC and SUF systems are principally interexchangeable, especially in an anaerobic environment. In E. coli and Thermotoga maritima, the suf gene cluster is arranged as sufA-sufB-sufC-sufD-sufS-sufE1 and sufC-sufB-sufD-sufS-sufU, respectively. In some hyperthermophilic archaea and bacteria, the SUF system has been proposed to be the sole pathway for cluster assembly. This implies that some components of the hyperthermophile SUF-related system might represent a primordial pathway for the Fe-S cluster biogenesis.
Although aerobic and anaerobic archaea produce numerous Fe-S proteins, the major components of the bacterial and eukaryal Fe-S cluster biosynthesis systems are not universally conserved in archaea. In S. tokodaii (and some other archaeal species), only the sufB, sufC, and sufD genes are conserved, which are arranged as the sufC(ST1201)-sufB(ST1200)-sufD(ST1199) gene cluster. SufB and SufD are paralogs and form a water-soluble, unorthodox ATP-binding cassette-like complex with SufC that has intrinsic ATPase activity. No sufA homolog could be identified in this archaeal genomic sequence. This is in line with a detection of archaeal SufBCD complex by the native proteome approach from native biomass using P. furiosus. Recently, the E. coli SufBC(2)D complex has been shown to function as a novel Fe-S scaffold machine and interacts with SufA for the Fe-S cluster transfer, and formation of the oxygen-labile [4Fe-4S] cluster was characterized by in vitro reconstitution of SufBC2D under anaerobic conditions. These findings strongly argue for the archaeal SufBC(2)D complex functioning as a possible Fe-S scaffold machine.
While SufA is absent in most archaea, the homologs of the bacterial apbC and eukaryotic NBP35 genes, coding for Fe-S cluster carrier proteins, are conserved in some archaea (ST0174 in the S. tokodaii genomic sequence). ApbC from Salmonella enterica is a homodimeric ATPase which can bind an Fe-S (presumably [4Fe-4S]) cluster and activate yeast apo-isopropylmalate dehydratase (apo-Leu1) in vitro, in an ATP-independent manner, and the S. enterica strains defective in apbC (mrp in E. coli) showed alterned thiamine biosynthesis and are impaired in Fe-S cluster metabolism. Likewise, the eukaryal ApbC homologs Cfd1 and Nbp35 form the extramitochondrial homotetrameric complex, and bind labile [4Fe-4S] clusters (after in vitro reconstitution), which can be transferred to target Fe-S apoproteins, but only when other CIA (cytosolic iron-sulfur protein assembly) proteins Nar1 and Cia1 co-exist. The archaeal ApbC/NBP35 homolog shows similar properties as S. enterica ApbC, and is a putative candidate for an Fe-S cluster shuttle that delivers a preassembled Fe-S cluster to a recipient apoprotein, although nothing is known to date about its interplay with the cognate SUF system. A missing piece in the SUF system of aerobic and thermoacidophilic archaea is a cysteine desulfurase (IscS/SufS/CsdA) homolog. A possibility is still open for novel cysteine desulfurases in these archaeal SUF systems. There are very few genetic and biochemical studies on the archaeal Fe-S cluster biosynthesis system so far, and further development of the genetic manipulation systems is needed to verify these hypotheses in a functional context.
up
Geometric tolerance of the cluster binding loop region and the thiophilicity with iron ions respect to the Fe-S cluster recognition
The de novo Fe-S cluster biosynthesis, which is catalyzed and regulated by a number of specific enzymes, can be divided into two major steps (Fig. 5). The first step is a transient de novo Fe-S cluster assembly on a scaffold protein requiring sulfur and iron donors. In the second step, the transient Fe-S cluster is dislocated from the scaffold protein, followed by transfer and insertion into recipient apoproteins, either during or shortly after the apoprotein generation and before the folding into its native-like conformation. A question of how the required (and rather ill-defined) binding site of a recipient protein-matrix, often categorized as the "binding motif" in the genome-wide bioinformatics, could select and bind a specific Fe-S cluster in the Fe-S protein biogenesis is considered below.
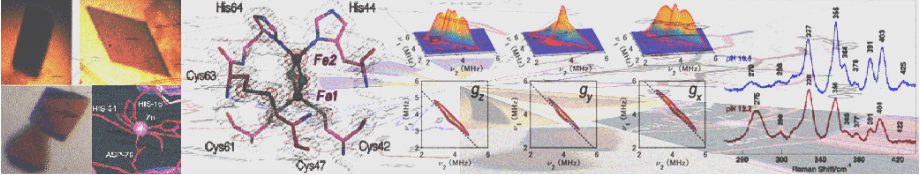
We used an archaeal Rieske-type [2Fe-2S] ferredoxin (called ARF) from Sulfolobus solfataricus P1 as a tractable model (Fig. 6). Rieske-type [2Fe-2S] clusters are ubiquitous in a variety of organisms, playing crucial electron transfer functions in respiratory chains, photosynthetic chains, and multicomponent oxygenase systems for biodegradation of aromatic and alkene compounds. In contrast to regular plant- and vertebrate-type [2Fe-2S] ferredoxins having complete cysteinyl ligations, the Rieske-type cluster has an asymmetric [2Fe-2S] core with the Sγ atom of each of the two cysteine residues coordinated to one iron site and the Nδ atom of each of the two histidine residues coordinated to the other iron site (e.g. PDB codes, 1rie, 1rfs, 1ndo, 1fqt, 1jm1, 1nyk and 2nuk.pdb). The structure of a bovine mitochondrial Rieske protein domain fragment suggests that its cluster-binding loops have a similar geometry to those found in the rubredoxin and zinc ribbon scaffolds.
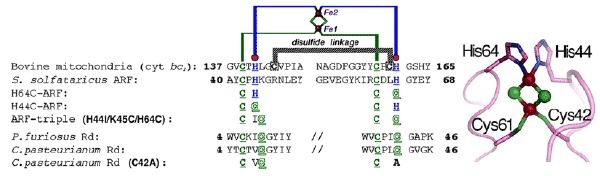
Figure 6. Multiple sequence alignment of the metal-binding sites of selected Rieske-type proteins and rubredoxins (Rd). The cluster-binding motif of S. solfataricus ARF is characteristic of Rieske-type ferredoxins involved in bacterial multicomponent oxygenases, containing two histidyl (blue) and two cysteinyl (green) ligands, and lacks two conserved cysteine residues (black) that serve as the disulfide linkage in respiratory Rieske proteins. The metal-binding motifs are underlined (left), and the structure of the cluster ligand residues of a bovine mitochondrial Rieske protein domain fragment (PDB code, 1rie.pdb) is shown, but with the S. solfataricus ARF numbering (right).
We have addressed the influence of substitution of each of the two outermost histidine ligands (His44 and His64) by cysteine on the properties of the Rieske-type [2Fe-2S] cluster in S. solfataricus ARF (Fig. 6). Replacement of one of the histidine ligands, His64, by cysteine allowed the assembly of a new low-potential [2Fe-2S] cluster with one histidine plus three cysteine ligands in the archaeal Rieske-type protein scaffold; whereas replacement of the other ligand, His44, by cysteine generated a protein that failed in cluster insertion and/or assembly (Fig. 7). Replacement of the two histidine ligands to the [2Fe-2S] cluster of S. solfataricus ARF by cysteine residues (in the H44C/H64C double mutant) largely impaired the cluster assembly in the recombinant variant protein (Fig. 8). In contrast, replacement of three residues (His-44, Lys-45, and His-64) in ARF by cysteines and isoleucine (H44I/K45C/H64C triple mutant), to mimic the mononuclear Fe(Cys)4 site in the P. furiosus rubredoxin, has allowed a rational design of the thermostable rubredoxin-like, mononuclear Fe(Cys)4 site in the recombinant ARF-triple mutant protein (Figs. 6, 8, 9).
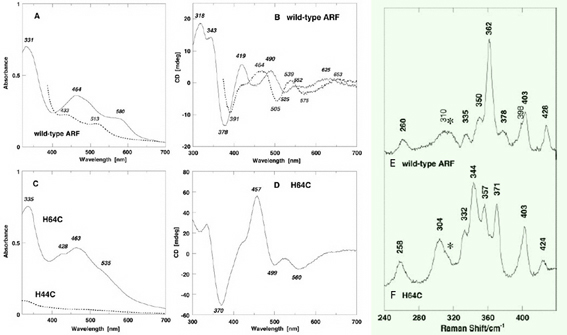
Figure 7. Visible-near UV absorption (A, C) and circular dichroism (CD) (B, D) spectra of the wild-type ARF (A and B) and the H64C variant (C and D) in the oxidized (solid) and dithionite-reduced (dashed) states. Cell length for visible-near UV CD spectra (B and D), 0.5 cm. mdeg, millidegrees; arb., arbitrary. Low temperature resonance Raman (RR) spectra of the oxidized [2Fe-2S] cluster with partial histidine ligation in the wild-type ARF (E) and the H64C variant (F). These RR spectra were obtained at 77 K using 488.0 nm Ar+ laser excitation. An asterisk indicates an ice mode from buffer. Our RR studies on ARF prove the extensive kinematic mixing of the Fe-N(imid) vibrations with the dominant Fe-S^b and/or Fe-S^t stretching characters of the immediate surroundings of a [2Fe-2S] cluster with partial histidine ligation in the 240–390 cm-1 region. Thus, the Fe-N(imid), Fe-S stretching vibrations and bond angle bending displacements are probably extensively coupled in the polypeptide backbone and widely spread into the molecule around the Rieske-type [2Fe-2S] cluster as reported for blue copper proteins and rubredoxin. Importantly the liganding histidine imidazole groups do not behave as simple point groups in the Rieske-type [2Fe-2S] system.
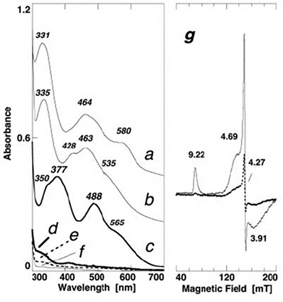
Figure 8. Comparative visible near-UV absorption spectra of the wild-type ARF (a) and the H64C (b), triple (H44I/K45C/H64C) (c), double (H44C/H64C) (d), H44C (e), H44I/K45C (f) variants, and X-band EPR spectra of the oxidized triple variant (g) at 4.3 (solid) and 12 K (dashed) (microwave power, 1.0 milliwatt; modulation amplitude, 0.63 mT; the g values are indicated in the figure). For the sake of clarity, offsets of 0.4 and 0.2 absorbance units have been applied to the spectra of the wild-type ARF (a) and H64C variant (b), respectively.
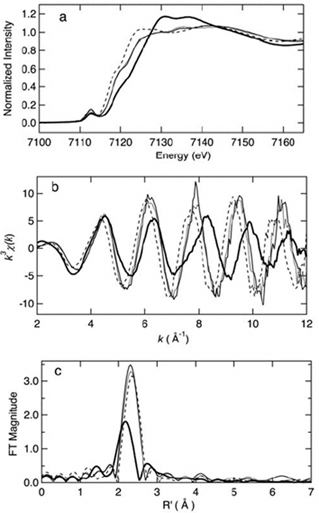
Figure 9. Iron K-edge x-ray absorption spectra edge spectra (a), k^3-weighted EXAFS (b), and sulfur phase-corrected Fourier transforms (FT) (c) of oxidized (thin solid) and reduced (dashed) iron-triple variant, oxidized Clostridium pasteurianum Fe-rubredoxin (dotted), and oxidized (thick solid) wild-type ARF. This figure was prepared by Dr. Ye Tao, Dr. Zhongrui Li, Dr. Jake Shokes, Dr. Nate Cosper, and Prof. Robert Scott (University of Georgia). Curve-fitting analyses are given in the original article of this work. The XAS data were collected at SSRL, which is operated by the US DOE, Division of Chemical Sciences (the SSRL Biotechnology program is supported by the NIH, Biomedical Resource Technology Program, Division of Research Resources).
These experiments demonstrate that the in vivo assembly of a [2Fe-2S] cluster in the Rieske protein scaffold is determined primarily by the nature and spacing of the ligands at the cluster binding loops which are often located near the protein surface in modular Fe-S protein. The two innermost cysteinyl ligand residues (Cys42 and Cys61) of S. solfataricus ARF are also essential for the cluster assembly and/or stability, suggesting that the thiophilicity of iron ions with the thiol-containing loop region is also important for the Fe-S cluster binding and/or stability. It seems plausible that a (kinetic) “native-like” semi-ordered structure of the cluster binding site in a folding intermediate may behave as a substrate in the enzyme-assisted [2Fe-2S] cluster assembly/maturation steps, where (i) the geometric tolerance of the metal-binding loops, allowed by the spacing and types of ligands near the protein surface, and (ii) the thiophilicity of iron ions with the thiol-containing loops should play decisive roles. This is in accord with the previous report by Meyer et al., showing the (unexpected) assembly of an oxidized [2Fe-2S] cluster into a recombinant, single-ligand-substituted (C42A) variant of Clostridium pasteurianum rubredoxin, whose polypeptide chain normally accommodates a mononuclear Fe(Cys)4 site in the wild-type protein (see Figs. 6, 8).
Although not experimentally tested, generality of this "geometrical tolerance plus thiophilicity" concept seems to also apply to the biogenesis of a cubane [4Fe-4S] cluster, considering also the established interconversion of the Fe-S cluster types (two [2Fe-2S] ↔ one [4Fe-4S]) on the IscU scaffold protein. Here the minimal requirement for the number of terminal cysteinyl ligands to a cubane [4Fe-4S] cluster is usually three in most simple and complex Fe-S proteins, and the fourth ligand at a (spatially) particular position can be an external ligand.
A likely biological and evolutionary benefit of having a polynuclear cluster site in a complex metalloenzyme would be that the cluster synthesis/assembly can be more strictly controlled by one or more specific synthesis-and-assembly apparatuses, thereby facilitating a unique redox chemistry for specific cellular needs—simple binding of a mononuclear transient metal site in a primordial metalloprotein might have been more severely influenced by the in vivo availability of environmental metal ions to the last universal common ancestors (due to the simpler metal binding equilibrium). Additionally, a cavity of sufficiently large size to accommodate a polynuclear cluster might reduce a potential problem of binding the wrong metal ion, correlated with the Irving-Williams series of the stability trend for aqueous metal-sulfur complexes in the order, Mn2+ < Fe2+ < Co2+ < Ni2+ < Cu2+ > Zn2+, even when diminishing iron levels posed a serious challenge for early aerobic archaea.
Prototypal polynuclear cluster formations, followed by early modular evolutionary events, afforded "stepwise" development of new catalytic and electron transfer functions of primordial complex metalloenzymes in biology. These enzymes consisted of ensembles of redox protein modules of convergent/divergent evolutionary origins, using a limited number of basic protein scaffolds, and could meet versatile requirements of early metabolisms and environmental conditions. Contemporary aerobic and thermoacidophilic archaea inherited the resultant intracellular Fe-S world from their anaerobic ancestors, and this world keeps running in an extraordinary environment by powering the enzyme-assisted Fe-S cluster biogenesis machinery.
*One can follow the link to read the original article of this work.
 Go to the ICC Project Details Page
Go to the ICC Project Details Page