 HOME
HOME
 Research Top
Research Top E. coli auxotrophic expression strains
E. coli auxotrophic expression strains 古細菌 Archaea | Sulfurisphaera tokodaii (f. Sulfolobus tokodaii)
古細菌 Archaea | Sulfurisphaera tokodaii (f. Sulfolobus tokodaii) モデル鉄硫黄蛋白質|Fe-S world in Sulfolobus
モデル鉄硫黄蛋白質|Fe-S world in Sulfolobus ISC-like ferredoxin (FdxB) from P. putida
ISC-like ferredoxin (FdxB) from P. putida Microbial "mitoNEET" homologs
Microbial "mitoNEET" homologs Archaeal respiratory complex II
Archaeal respiratory complex II 複合金属酵素の発現
複合金属酵素の発現 Ligand mutagenesis
Ligand mutagenesis 鉄硫黄酵素の分光学
鉄硫黄酵素の分光学 金属コファクターの分子進化
金属コファクターの分子進化 ICC Project
ICC Project
日本医科大学、医学部、生化学・分子生物学(代謝・栄養学)所属の 岩崎 俊雄グループでは、好熱菌等を主材料とした各種金属酵素の構造機能進化や、アミノ酸要求性の新規大腸菌発現宿主株作成につき、国内外の様々な研究機関と共同研究を実施しています。
 Publication List
Publication List Go to the Research Top Page
Go to the Research Top Page(Site-directed) "Ligand" mutagenesis
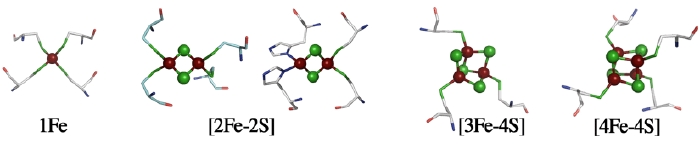
The structure of a metal site in metalloenzymes critically influences the fine-tuning of redox and/or catalytic activities in biology. The substitution and/or displacement events at the local metal-binding site(s) in a protein might have greatly enhanced their capabilities of conducting a wide range of unique redox chemistry in biological electron transfer conduits which often use a limited number of basic protein scaffold. Iron-sulfur (Fe-S) cluster prosthetic groups, consisting of nonheme iron and acid-labile inorganic sulfide atoms, are functionally highly versatile and may be among the most ancient modular metallo-cofactors. The general importance of these Fe-S cofactors in biology is largely attributable to specific features of iron and sulfur chemistry, and the assembly and interplay of the Fe-S cluster core with the surrounding protein is the key to in-depth understanding of the underlying mechanisms.
Ligand mutagenesis studies on hyperthermophilic archaeal Rieske-type [2Fe-2S] proteins
Among the biological Fe-S clusters with at least one noncysteinyl ligand, “Rieske-type” [2Fe-2S] clusters are ubiquitous in a variety of organisms, playing crucial electron transfer functions in respiratory chains, photosynthetic chains, and multicomponent oxygenase systems for biodegradation of aromatic and alkene compounds. In contrast to regular plant- and vertebrate-type ferredoxins having complete cysteinyl ligations, the Rieske-type cluster has an asymmetric [2Fe-2S] core with the Sγ atom of each of the two cysteine residues coordinated to one iron site and the Nδ atom of each of the two histidine residues coordinated to the other iron site (e.g. PDB codes, 1rie, 1rfs, 1ndo, 1fqt, 1jm1, 1nyk and 2nuk.pdb). The crystal structure of a mitochondrial Rieske protein domain fragment suggests that its cluster-binding loops have similar geometry as those found in the rubredoxin (Rd) and zinc ribbon scaffolds. The presence of the two histidine ligands to the Rieske centers is considered to be essential in respiratory cytochrome bc complexes in which the protonation state of the histidine ligands plays a crucial role in the quinol-oxidizing Qo-site turnover.
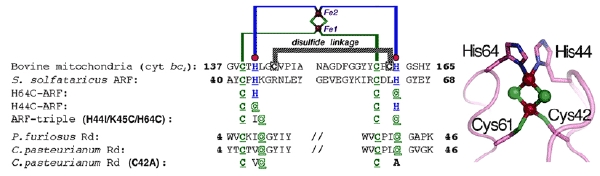
Figure 1. Multiple sequence alignment of the metal-binding sites of selected Rieske-type proteins and rubredoxins (Rd). The cluster-binding motif of S. solfataricus ARF is characteristic of Rieske-type ferredoxins involved in bacterial multicomponent oxygenases, containing two histidyl (blue) and two cysteinyl (green) ligands, and lacks two conserved cysteine residues (black) that serve as the disulfide linkage in respiratory Rieske proteins. The metal-binding motifs are underlined (left), and the structure of the cluster ligand residues of a bovine mitochondrial Rieske protein domain fragment (PDB code, 1rie.pdb) is shown, but with the S. solfataricus ARF numbering (right).
We have addressed the influence of substitution of each of the two outermost histidine ligands (His44 and His64) by cysteine on the properties of the Rieske-type [2Fe-2S] cluster in the hyperthermophilic archaeal Rieske-type [2Fe-2S] ferredoxin (called ARF) from Sulfolobus solfataricus P1 as a tractable model (Fig. 1). Replacement of one of the histidine ligands, His64, by cysteine allowed the assembly of a new low-potential [2Fe-2S] cluster with one histidine plus three cysteine ligands in the archaeal Rieske-type protein scaffold; whereas replacement of the other ligand, His44, by cysteine generated a protein that failed in cluster insertion and/or assembly (Fig. 2). Replacement of the two histidine ligands to the [2Fe-2S] cluster of S. solfataricus ARF by cysteine residues (in the H44C/H64C double mutant) largely impaired the cluster assembly in the recombinant variant protein (Fig. 3). Thus, while two cysteine ligand residues (Cys42 and Cys61) are essential for the cluster assembly and/or stability, the contributions of the two histidine ligands to the cluster assembly in the archaeal Rieske-type ferredoxin appear to be inequivalent as indicated by much higher stability of the His64 → Cys variant (H64C) than the His44 → Cys variant (H44C).
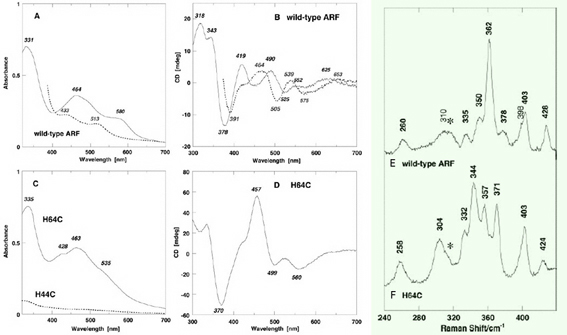
Figure 2. Visible-near UV absorption (A, C) and circular dichroism (CD) (B, D) spectra of the wild-type ARF (A and B) and the H64C variant (C and D) in the oxidized (solid) and dithionite-reduced (dashed) states. Cell length for visible-near UV CD spectra (B and D), 0.5 cm. mdeg, millidegrees; arb., arbitrary. Low temperature resonance Raman (RR) spectra of the oxidized [2Fe-2S] cluster with partial histidine ligation in the wild-type ARF (E) and the H64C variant (F). These RR spectra were obtained at 77 K using 488.0 nm Ar+ laser excitation. An asterisk indicates an ice mode from buffer. Our RR studies on ARF prove the extensive kinematic mixing of the Fe-N(imid) vibrations with the dominant Fe-S^b and/or Fe-S^t stretching characters of the immediate surroundings of a [2Fe-2S] cluster with partial histidine ligation in the 240–390 cm-1 region. Thus, the Fe-N(imid), Fe-S stretching vibrations and bond angle bending displacements are probably extensively coupled in the polypeptide backbone and widely spread into the molecule around the Rieske-type [2Fe-2S] cluster as reported for blue copper proteins and rubredoxin. Importantly the liganding histidine imidazole groups do not behave as simple point groups in the Rieske-type [2Fe-2S] system.
In contrast, replacement of three residues (His-44, Lys-45, and His-64) in ARF by cysteines and isoleucine (H44I/K45C/H64C triple mutant), to mimic the mononuclear Fe(Cys)4 site in the Pyrococcus furiosus rubredoxin, has allowed a rational design of the thermostable rubredoxin-like, mononuclear Fe(Cys)4 site in the recombinant ARF-triple mutant protein (Figs. 1, 3, 4).
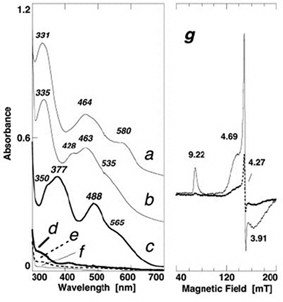
Figure 3. Comparative visible near-UV absorption spectra of the wild-type ARF (a) and the H64C (b), triple (H44I/K45C/H64C) (c), double (H44C/H64C) (d), H44C (e), H44I/K45C (f) variants, and X-band EPR spectra of the oxidized triple variant (g) at 4.3 (solid) and 12 K (dashed) (microwave power, 1.0 milliwatt; modulation amplitude, 0.63 mT; the g values are indicated in the figure). For the sake of clarity, offsets of 0.4 and 0.2 absorbance units have been applied to the spectra of the wild-type ARF (a) and H64C variant (b), respectively.
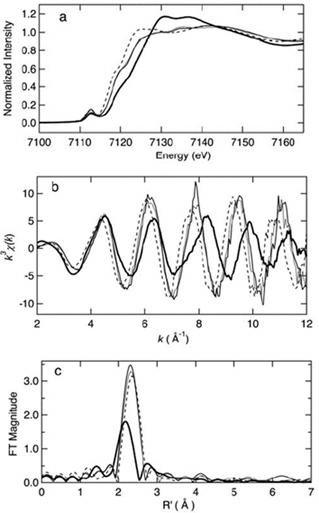
Figure 4. Iron K-edge x-ray absorption spectra edge spectra (a), k^3-weighted EXAFS (b), and sulfur phase-corrected Fourier transforms (FT) (c) of oxidized (thin solid) and reduced (dashed) iron-triple variant, oxidized Clostridium pasteurianum Fe-rubredoxin (dotted), and oxidized (thick solid) wild-type ARF. This figure was prepared by Dr. Ye Tao, Dr. Zhongrui Li, Dr. Jake Shokes, Dr. Nate Cosper, and Prof. Robert Scott (University of Georgia). Curve-fitting analyses are given in the original article of this work. The XAS data were collected at SSRL, which is operated by the US DOE, Division of Chemical Sciences (the SSRL Biotechnology program is supported by the NIH, Biomedical Resource Technology Program, Division of Research Resources).
Our successful rational design of the thermostable Rd-like, mononuclear iron site with complete cysteinyl ligation in the recombinant triple mutant protein (Figs. 1–4) experimentally shows that the in vivo assembly of a [2Fe-2S] cluster in the Rieske protein scaffold is determined by the nature and spacing of the ligands at the cluster-binding motif (Figs. 1, 3). It seems plausible to postulate that a “native-like” semi-ordered structure of the cluster binding site in a folding intermediate, wherein the spacing and types of ligands near the protein surface should play a decisive role, may behave as a substrate in the enzyme-assisted dinuclear cluster assembly/maturation steps. This is in accord with the previous report by Meyer et al., clearly showing the (unexpected) assembly of an oxidized [2Fe-2S] cluster into a recombinant, single ligand-substituted (C42A) variant of Clostridium pasteurianum Rd, whose polypeptide chain normally accommodates a mononuclear Fe(Cys)4 site in the wild-type protein. In conjunction with the results of our work showing negligible assembly of a [2Fe-2S] cluster in the ARF H44C variant (in contrast to the H64C variant), we suggest that His-44 in the cluster-binding loop (Fig. 1) plays a crucial role in the cluster assembly and metal-type recognition in the archaeal Rieske-type protein scaffold.
The “two cysteines plus two histidines”-type coordination pattern commonly observed in the Rieske-type proteins/domains might have been preferred in the modular evolution process of an early Rieske-type protein scaffold, as a consequence of the geometric tolerance of the metal-binding loops for the [2Fe-2S] cluster insertion and/or assembly. The ligand exchange and metal- and/or cluster-type conversion might have been key evolutionary events from an archetypal mononuclear metalloprotein module toward a modern, high-potential Rieske protein subunit of the respiratory complexes III, in which one of two histidyl ligands plays crucial electron/proton transfer roles in the quinol-oxidizing Qo-site catalysis. Likewise, the dinuclear copper center in the CuA site of mitochondrial and bacterial respiratory complexes IV, which is the primary electron acceptor site from mobile cytochrome c, is known to be structurally and evolutionarily related to the mononuclear copper proteins such as azurin and plastocyanin in the photosynthetic electron transfer chain. Possible common prototypal evolutionary patterns in both cases are (i) the mono- to di-nuclear metal center conversion with a few residue substitutions in the immediate metal-binding site and (ii) the modular evolution from water-soluble (mononucleartype) ancestral protein modules, with minimal changes of the native protein scaffolds. This evolutionary pattern can explain the complicated quaternary structural features of a wide range of redox metalloenzyme complexes, yet consists of a relatively limited number of protein scaffolds within a variety of biological electron transfer pathways. It is envisaged that the evolutionary origins of some primordial redox protein modules may have preceded the divergence of the three different domains of life.
A likely biological and evolutionary benefit of having a polynuclear cluster site in a complex metalloenzyme would be that the cluster synthesis/assembly can be more strictly controlled by one or more specific synthetic and assembly apparatuses thereby facilitating a unique redox chemistry for specific cellular needs; the simple binding of a mononuclear transient metal site in a primordial metalloprotein might have been more severely influenced by the in vivo availability of environmental metal ions to the common ancestral organisms (because of the simpler metal binding equilibrium). We suggest that prototypal polynuclear cluster formations, followed by early modular evolutionary events, might have afforded a “stepwise” development of new catalytic and electron transfer functions of primordial complex metalloenzymes, consisting of ensembles of redox protein modules of convergent/divergent evolutionary origins using a limited number of basic protein scaffolds to meet the versatile requirements of early metabolisms and environmental conditions.
*One can follow the link to read the original article of this work.
up
Crystallization of a hyperthermophilic archaeal Rieske protein variant (SDX-triple) with an engineered rubredoxin-like mononuclear iron site
Our site-directed mutagenesis studies have indicated the importance of types and spacing of terminal ligands in the in vivo cluster recognition/insertion/assembly in metallosulfur protein scaffolds. By mimicking the mononuclear iron site in the Pyrococcus furiosus Rd involved in the molecular oxygenscavenging system, replacement of three residues (His44, Lys45 and His64) in Sulfolobus solfataricus ARF by cysteines and isoleucine (ARF-triple; H44I/K45C/H64C) resulted in an Rd-type mononuclear Fe(Cys)4 site in the Rieske-type protein scaffold (Figs. 1, 3, 4). A deeper understanding of the metal-binding site design and evolutionary divergency from the same protein template to promote new and specific functionalities would require a knowledge of structural information at atomic resolution, but no suitable crystals of ARF-triple have been produced.
Among the Rieske-type [2Fe–2S] proteins characterized so far, archaeal sulredoxin (SDX) from the hyperthermoacidophile Sulfolobus tokodaii strain 7 (DDBJ-EMBL-GenBank accession No. AB023295) is the closest homologue of the S. solfataricus ARF. This 12 kDa protein is unusual in that it is also weakly homologous to the extrinsic cluster-binding domain of cytochrome bc-associated Rieske proteins with a solvent-exposed consensus disulfide linkage (Fig. 5A), despite the inherent absence of the transmembrane domain. Native and recombinant SDX have been crystallized, and the crystal structures were recently solved at 1.15-Å and 2.0-Å resolution, respectively [to be published]. In place of the archaeal Rieske [2Fe-2S] cluster, an archetypal mononuclear iron site has rationally been designed into S. tokodaii SDX, by three residue replacements with reference to the P. furiosus rubredoxin sequence (Fig. 5A,B). The resulting SDX variant, SDX-triple (H44I/A45C/H64C), has been purified and crystallized by the hanging-drop vapor-diffusion method using 65% (v/v) 2-methyl-2,4-pentanediol, 0.025 M citric acid and 0.075 M sodium acetate trihydrate pH 4.3 (Fig. 5C). The crystals diffract to 1.63-Å resolution and belong to the triclinic space group P1, with unit-cell parameters a = 43.56, b = 76.54, c = 80.28 Å, α = 88.12, β = 78.82, γ = 73.46°. The asymmetric unit contains eight protein molecules.
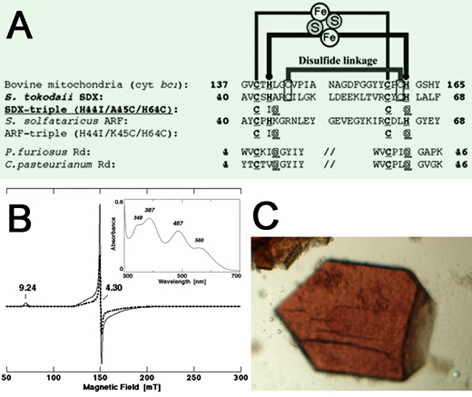
Figure 5. (A) Multiple sequence alignment of the metal binding sites of selected Rieske proteins and Rds. The cluster binding motif of S. tokodaii SDX is characteristic of the high-potential Rieske protein family and has two conserved cysteine residues that serve as the solvent exposed disulfide linkage (boxed). Accession numbers: Bovine mitochondrial cytochrome bc1-associated, Rieske protein fragment, P13272; Sulfolobus tokodaii SDX, AB023295; Sulfolobus solfataricus ARF (hypothetical ORF c06009), CAA669492, AB047031; Pyrococcus furiosus Rd, P24297; Clostridium pasteurianum Rd, P00268. The metal binding motifs are underlined. (B) X-band EPR spectra at 4.2 (solid) and 12.1 K (dashed) of the oxidized SDX-triple (H44I/A45C/H64C) variant, measured with the following settings: microwave power, 1.0 mW; modulation amplitude, 0.2 mT; the g values are indicated in the figure. It displays a resonance at g = 9.24 associated with one principal direction of the lowest Kramers doublet (±1/2 or ±5/2) at 4.2 K and a number of features in the g ≈ 3.8-4.7 range associated with the three principal directions of the ±3/2 doublet. These features are very similar to those reported for archaeal and bacterial Rds, and clearly show the presence of a high-spin ferric iron site. Inset shows the visible-near UV absorption spectrum of the purified SDX-triple variant recorded at room temperature. (C) Typical crystals of the SDX-triple variant. The maximum dimensions of the triclinic crystals are approximately 0.2 x 0.2 x 0.05 mm.
*One can follow the link to read the original article of this work.
up
Crystal structure of the archaeal Rieske protein variant (SDX-triple) with an engineered rubredoxin-like mononuclear iron site
Sequence motif-specific assignment of two [2Fe-2S] clusters in mammalian xanthine oxidoreductase studied by site-directed mutagenesis
Xanthine oxidoreductase (XOR), xanthine dehydrogenase (XDH, EC 1.1.1.204) and xanthine oxidase (XO, EC 1.2.3.2), catalyzes the oxidation of xanthine to uric acid with concomitant reduction of NAD+ or molecular oxygen. It is a homodimer of molecular weight 300,000, with each subunit having one non-covalently bound FAD, one molybdopterin-bound mononuclear molybdenum (Mo) center, and two [2Fe-2S]2+,1+ clusters (Fig. 6), and is the most extensively characterized member of the mononuclear molybdenum (Mo)-containing hydroxylase family. The reduction and oxidation reactions of the catalytic cycle of XOR are spatially separated: oxidative hydroxylation of xanthine to uric acid takes place at the Mo center in the 90-kDa molybdenum domain, and reducing equivalents thus introduced into enzymes are transferred rapidly via intramolecular electron transfer to FAD in the 40-kDa flavin domain, where physiological oxidation occurs. For this reason, intramolecular electron transfer between the Mo center and FAD center is an integral aspect of the overall catalytic sequence of this complex flavo-metalloprotein.
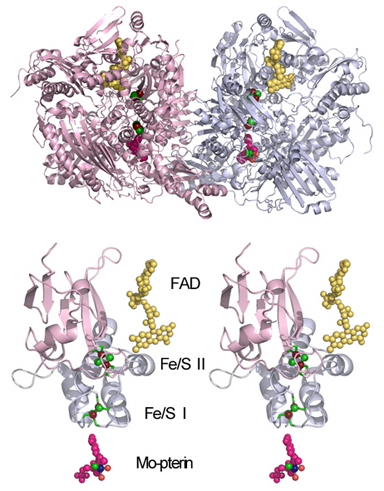
Figure 6. Structure of bovine milk XOR (PDB entry, 1FO4.pdb) (top) and its Fe/S domain highlighting the cofactor arrangement (in a wall-eye stereo view, bottom).
The two [2Fe-2S] clusters (Fe/S I and II) in the 20-kDa iron-sulfur (Fe/S) domain of XOR (Fig. 6, bottom) are indistinguishable in terms of the visible absorption spectra, but the midpoint redox potential of the Fe/S II center is ~80 mV more positive than that of the Fe/S I center and their electron paramagnetic resonance (EPR) properties can be distinguished by their different g values and their different saturation properties at various temperatures. The Fe/S I center exhibits a rhombic EPR signal (g = 2.02, 1.93, and 1.90 ± 0.01), which is similar to those observed in the regular plant-type [2Fe-2S] ferredoxins and is readily observable at temperatures up to 40 K. On the other hand, the Fe/S II center exhibits an unusually broad EPR signal (g = 2.10 ± 0.02, 1.98 ± 0.015, and 1.91 ± 0.01) which is characteristic of some Mo-containing hydroxylases and can be observed only below 22 K. These EPR signals can be commonly observed in XOR and the related mononuclear Mo-containing hydroxylases with two [2Fe-2S] clusters.
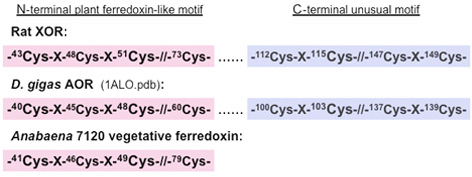
Figure 7. Schematic representation of the two [2Fe-2S] cluster binding motifs in the Fe/S domain of rat XOR and D. gigas AOR (PDB entry, 1ALO.pdb), and the plant-type ferredoxin motif of Anabaena 7120 vegetative ferredoxin. Only conserved and potential cysteine ligand residues are shown.
The primary structures of the Fe/S domain of some mononuclear Mo-containing hydroxylases such as XOR (Fig. 6, bottom) and Desulfovibrio gigas aldehyde oxidoreductase (AOR) show the presence of eight strictly conserved cysteine residues (Fig. 7). These residues have been shown to serve as ligands to the two [2Fe-2S] clusters in the corresponding crystal structures. The N-terminal half of the Fe/S domain (Fig. 6, bottom, pink ribbon) contains four of them that are arranged to form the canonical [2Fe-2S] cluster binding motif of the regular plant-type ferredoxins (Fig. 7). The C-terminal half (Fig. 6, bottom, blue ribbon) contains the other four that are arranged in the unusual -Cys-Xaa2-Cys-//-Cys-Xaa1-Cys- motif in a unique protein fold which is uniquely found in this enzyme family (Fig. 7).
Although the sequence motif-specific assignment of the two [2Fe-2S] clusters to the corresponding EPR signals would greatly enhance our understanding of the structure-function relation in XOR, this remained controversial and was not conclusively established when this study was undertaken. One possible approach to elucidate this problem is the combined application of site-directed mutagenesis and EPR spectroscopy, as has been successfully utilized for assignment of potential ligand residues to Fe/S cluster(s) in some Fe/S proteins with unknown structure and for analysis of the modified spectroscopic and/or redox properties of a particular redox site in some metalloproteins. In this work, we unambiguously establish the sequence motif-specific assignment of the two [2Fe-2S] clusters in recombinant rat liver XOR, taking advantage of the baculovirus-insect cell (Sf9) expression system described by Nishino et al.
The recombinant rat liver XOR produced by using the baculovirus-insect cell (Sf9) expression system was a heterogeneous mixture as the molybdo (native) dimeric, demolybdo dimeric and demolybdo monomeric forms. Purification and the properties of each form of the recombinant XOR has been described elsewhere. Although the molybdo XOR can be obtained after the folate affinity column chromatography, the amount and the stability of the recombinant wild-type and mutant enzymes are not enough to permit the usage of the molybdo form in EPR experiments. In the following EPR analysis, we utilized the purified demolybdo or partially purified mixture in some experiments. This does not interfere the sequence motif-specific assignment of the two [2Fe-2S] clusters in XOR as described below.
up
EPR spectra of the recombinant wild-type XOR dimer
Figure 8 shows the EPR spectra of dithionite-reduced molybdo and demolybdo dimeric XOR recorded at 16-39 K. The native molybdo dimeric XOR prepared from rat liver (traces A and B) exhibited a rhombic EPR signal at g = 2.02, 1.93, and 1.90 attributed to the Fe/S I center (observable at 39 K), in addition to overlapping broad rhombic resonance at g = 2.11, ~2.00, and ~1.90 attributed to the Fe/S II center. These EPR properties are essentially identical to those reported for milk XOR.
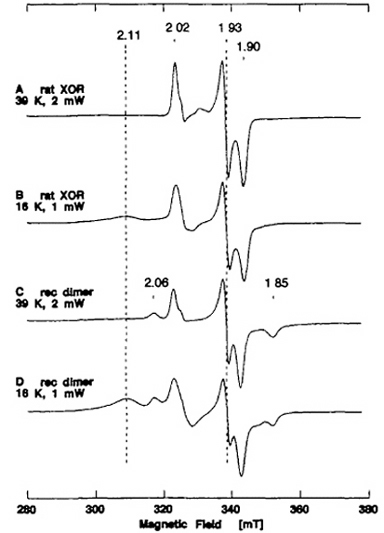
Figure 8. EPR spectra of the dithionite-reduced form of native XOR from rat liver (A, B) and recombinant wild-type XOR dimer (C, D). The purified recombinant wild-type demolybdo dimer of XOR contained ~8Fe/2FAD (mol/mol), indicating the presence of two [2Fe-2S] clusters per protomer, and showed no activity with xanthine and DCPIP as substrates, due to the absence of the Mo-pterin center. Instrument settings for the X-band EPR spectroscopy: the temperature and microwave power are indicated in the figure; modulation amplitude, 0.79 millitesla. The g values are indicated in the figure.
The purified recombinant wild-type demolybdo dimer of XOR elicited broad new resonances at g ~ 2.06 and 1.85, in addition to the signals attributed to the Fe/S I and II centers (Fig. 8C, D). The relaxation behavior of the new g ~ 2.06 and 1.85 resonances is similar to that of the S = 1/2 EPR signal attributed to the Fe/S I center (Fig. 8A, C), and these signals can be observed at 39 K where the EPR signal attributed to the Fe/S II center is saturated and not observable. This suggests that the additional EPR features observed in the recombinant demolybdo dimeric enzyme cannot be attributed to the splitting of the EPR signal of the Fe/S I center due to magnetic interaction between the Fe/S I and II centers. Preliminary simulation and spin integrations also suggested that the g = 2.02 signal and the g = 2.06 signal are present approximately in a 1:1 ratio at 16 K and 39 K (data not shown). These results indicate that the Fe/S I site in the recombinant demolybdo dimeric enzyme is heterogeneous due to the absence of the Mo-pterin center and probably exists in (at least) two different forms and/or states in a ~1:1 ratio, whereas the Fe/S II site remains homogenous. The same observation has been reported with the fully demolybdo form of bovine milk XO. We suggest that the presence of the g = 2.02 signal and the additional g = 2.06 signal in a ~1:1 ratio is characteristic of demolybdo-dimeric XOR. Although the EPR signal of the Fe/S II center interferes the g = 2.06 signal at lower temperatures, the g = 2.02 signal of the recombinant wild-type demolybdo dimer is apparently more intense in the first-derivative spectrum, and is therefore used as a diagnostic of the Fe/S I center in the following analysis.
up
EPR properties of C115S mutant enzyme
In order to assign the Fe/S I and II centers in the recombinant demolybdo dimeric XOR, a mutant enzyme C115S, having a single amino acid replacement of Cys-115 by Ser (see Fig. 7), was constructed and expressed by using the baculovirus-insect cell expression system. The dimeric form of the resultant mutant enzyme C115S was rather unstable during purification under auxogenic conditions. Hence, the EPR spectra of dithionite-reduced C115S were recorded shortly after fractionation of the recombinant enzyme from the cell lysate by DEAE cellulose column chromatography (Fig. 9C-F). The control EPR experiment with the reduced recombinant wild-type enzyme from the same purification step confirmed little interference from other paramagnetic contaminants from the host insect cells in the g = 2 region under the applied conditions (data not shown).
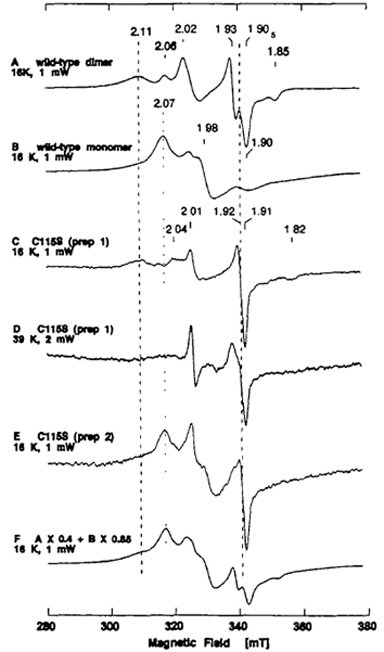
Figure 9. EPR spectra of the dithionite-reduced form of recombinant wild-type dimer (A) and monomer at 16 K (B), and partially purified C115S mutant enzyme at 16 K (C, E) and 39 K (D). The near-axial EPR signal at g = 1.92 in the spectra C-E cannot be simulated as the sum of the spectra of the recombinant wild-type dimer and monomer (F). The EPR properties of recombinant wild-type dimer (A) and monomer at 16 K (B) are substantially different, and the details of the properties of the recombinant wild-type monomer have been reported elsewhere. The partially purified C115S was a mixture of monomeric and dimeric forms with a typical ratio of A450 nm and A550 nm of ~4.7 (varied from preparation to preparation). The negligible activity/flavin ratio (AFR) value (0.1) and specific xanthine-DCPIP oxidoreductase activity of 2.8-5.1 mol/min/mol of FAD (less than 0.5-1% of native enzyme) suggested that it is predominantly the demolybdo form. Instrument settings for the EPR spectroscopy: microwave power, 1 mW for A, B, C, E, and 2 mW for D; modulation amplitude, 0.79 millitesla. The g values are indicated in the figure.
EPR analysis of several different batches of partially purified C115S mutant enzyme in the reduced state showed that every batch contained a novel [2Fe-2S]1+ center with near axial symmetry centered at g = 1.92 (Fig. 9C, E), in addition to the broad EPR signal characteristic of the Fe/S II center at g = 2.11 that is indistinguishable from that of the recombinant wild-type dimeric enzyme (Fig. 9A, C). The near-axial EPR signal at g = 1.92 is considerably broadened at 39 K (Fig. 9D). On the other hand, no rhombic EPR signal at g = 2.02, 1.93, 1.90 (the average g-factor, gav = 1.95) characteristic of the Fe/S I center of the wild-type dimer (Fig. 9A) could be detected in any C115S preparation examined (Fig. 9C-E). These data suggest that Cys-115 is one of the cysteine ligands to the Fe/S I center in the dimeric enzyme, and that the serine residue (Ser-115) serves as a non-cysteinyl ligand to this [2Fe-2S] cluster in the mutant enzyme C115S (Fig. 9C-E). A similar EPR spectral change has been reported for the Cys-49 → Ser mutation of the Anabaena 7120 vegetative ferredoxin, which also shows the near-axial EPR signal similar to those of the vertebrate-type ferredoxins and affects primarily the Fe(III) site of the [2Fe-2S] cluster. It should be noted, however, that Cys-115 of rat XOR is located in the unusual [2Fe-2S] cluster binding motif, -Cys-Xaa2-Cys-//-Cys-Xaa1-Cys-, at the C-terminal part of the Fe/S domain, with unique folding (Fig. 6, bottom, blue ribbon, and Fig. 7).
up
EPR properties of C51S mutant enzyme
The N-terminal part of the Fe/S domain (Fig. 6, bottom, pink ribbon) has the -Cys-Xaa4-Cys-Xaa2-Cys-//-Cys- motif which is characteristically observed in regular plant-type ferredoxins (Fig. 7). A mutant enzyme C51S, having a single amino acid replacement of Cys-51 in the N-terminal motif with serine, was constructed and expressed by using the baculovirus-insect cell expression system. As in the case reported for C115S, the resulting mutant enzyme C51S was rather unstable during purification under auxogenic conditions. Hence, the EPR spectra of dithionite-reduced C51S were recorded shortly after fractionation of the mutant enzyme from the cell lysate by DEAE cellulose column chromatography (Fig. 10). The EPR signals attributed to the [2Fe-2S]1+ clusters in partially purified C51S are complicated due to the presence of both dimeric and monomeric forms (Fig. 10C, D), and are very similar to the sum of the spectra of the recombinant wild-type dimer and monomer (Fig. 10B).
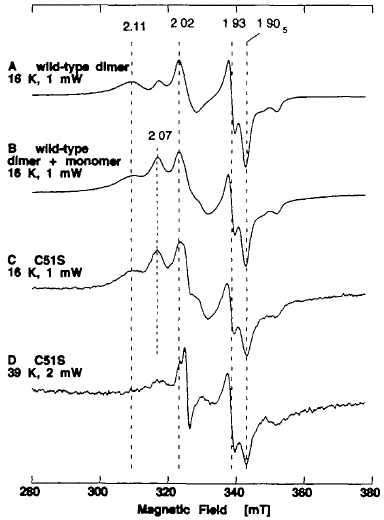
Figure 10. EPR spectra of the dithionite-reduced form of recombinant wild-type dimer at 16 K (A), and partially purified C51S mutant enzyme at 16 K (C) and 39 K (D). The EPR spectrum C could be simulated as the sum of the spectra of the wild-type dimer and monomer (B). The partially purified C51S was a mixture of monomeric and dimeric forms with a typical ratio of A450 nm and A550 nm of ~4.4. The AFR value of 1.6 and the negligible xanthine-DCPIP oxidoreductase activity (2.8 mol/min/mol of FAD; less than 0.5% of native enzyme) suggested that it is predominantly the demolybdo form. Instrument settings for the EPR spectroscopy: microwave power, 1 mW for A-C, and 2 mW for D; modulation amplitude, 0.79 millitesla. The g values are indicated in the figure.
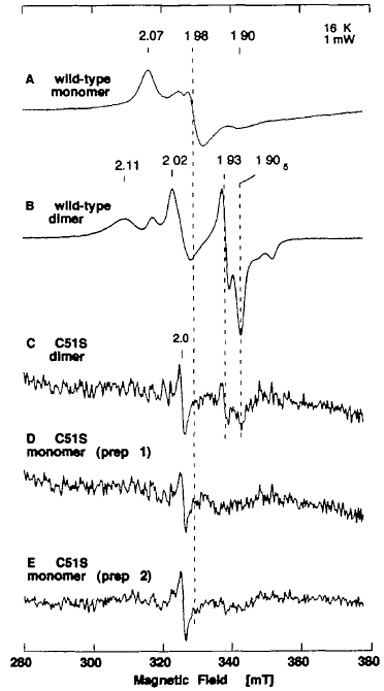
Figure 11. EPR spectra of the dithionite-reduced form of recombinant wild-type monomer (A), wild-type dimer (B), purified C51S dimer (C), and purified C51S monomer (D, E). Purified C51S mutant enzyme preparations gave EPR spectra with a relatively poor signal-to-noise ratio, because of the instability of the bound [2Fe-2S] cluster after purification. Instrument settings for the EPR spectroscopy: temperature 16 K; microwave power, 1 mW; modulation amplitude, 0.79 millitesla. The g values are indicated in the figure.
Although no change in the EPR signal attributed to Fe/S I center was observed in crude C51S at 16 K and 39 K (Fig. 10C, D), the resulting mutant enzyme was more unstable than the recombinant wild-type enzyme, as indicated by faster bleaching of the brown color (data not shown). As opposed to the wild-type enzyme (Fig. 11A, B), further purification of crude C51S preparations under aerobic conditions resulted in marked loss of the [2Fe-2S] clusters in both the dimeric and monomeric forms, giving rather featureless EPR signals with a very poor signal-to-noise ratio (Fig. 11C-E). Because the Cys → Ser mutants of Anabaena 7120 vegetative ferredoxin have been reported to be less stable than the wild-type ferredoxin (as expected from the relative pKa values of cysteine and serine), the instability of the clusters in C51S is likely to be due to the single amino acid replacement of Cys-51 by serine, which serves as a non-cysteinyl ligand to the [2Fe-2S] cluster (i.e., the Fe/S II center) in partially purified C51S (Figs. 10, 11).
We also constructed and expressed the C51A mutant enzyme, having a single amino acid replacement of Cys-51 with alanine, by using the baculovirus-insect cell expression system. Although the expression of the recombinant mutant enzyme was confirmed by western blot analysis, the product was mostly obtained as an insoluble material (data not shown). This suggests that Cys-51 is a ligand residue with structural importance in protein conformation and/or folding.
up
EPR properties of C43S mutant enzyme
In contrast to the C51S and C51A mutant enzymes described above, the mutant enzyme C43S, which has a single amino acid replacement of Cys-43 with serine in the N-terminal plant ferredoxin-like -Cys-Xaa4-Cys-Xaa2-Cys-//-Cys- motif (Fig. 6, bottom, pink ribbon, and Fig. 7), was sufficiently stable to allow separation of the dimeric and monomeric forms from each other by gel filtration column chromatography. The EPR spectra of the dimeric and monomeric forms of the dithionite-reduced C43S were substantially different from those of the recombinant wild-type demolybdo XOR (Fig. 12). The EPR spectra of the C43S dimer at 8-60 K clearly showed the presence of two overlapping S = 1/2 [2Fe-2S]1+ clusters with different relaxation behaviors (Fig. 13). The EPR lineshape attributed to the Fe/S I center in C43S was essentially indistinguishable from that in the recombinant wild-type dimer (gav = 1.95) (Fig. 13A, E), whereas the signal attributed to the Fe/S II center was markedly modified, exhibiting a near-axial type signal at g = 2.05, ~1.94, ~1.93 (gav ~ 1.97) (Fig. 13F). These data showed that Ser-43 serves as a non-cysteinyl ligand to the Fe/S II center in the plant-type ferredoxin subdomain in the C43S dimer, without significant modification of the EPR lineshape attributed to the Fe/S I center. Interestingly, the effect of replacement of Cys-43 → Ser in XOR resulting the reduced Fe/S II center with gav of ~1.97 is considerably different from that of the equivalent mutation (Cys-41 → Ser) in reduced Anabaena 7120 vegetative ferredoxin resulting the cluster with the lower gav of ~ 1.92.
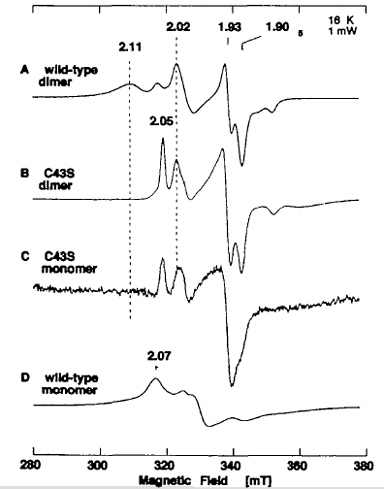
Figure 12. EPR spectra of the dithionite-reduced form of recombinant wild-type dimer (A), dimeric C43S mutant enzyme (B), monomeric C43S mutant enzyme (C), and recombinant wild-type monomer (D). The dimeric form of C43S had a typical ratio of A450 nm and A550 nm of ~3.8 (estimated to contain ~1.8 [2Fe-2S] centers per FAD (mol/mol)), with the AFR value of ~2 and the negligible xanthine-DCPIP oxidoreductase activity of 3.1 mol/min/mol of FAD (0.6% of native enzyme), suggesting predominantly the demolybdo form. The monomeric form of C43S showed no activity with xanthine and DCPIP as substrates, as in the case of the wild-type demolybdo-monomeric XOR. Instrument settings for the EPR spectroscopy: temperature 16 K; microwave power, 1 mW; modulation amplitude, 0.79 millitesla. The g values are indicated in the figure.
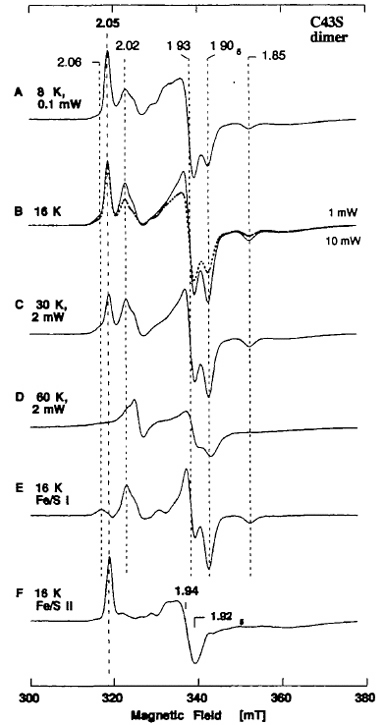
Figure 13. EPR spectra of the dithionite-reduced form of purified dimeric C43S mutant enzyme at 8-60 K. The spectra E and F were obtained by subtraction of the EPR spectra at 16 K recorded at the microwave power of 1 mW (B, solid trace) and 10 mW (B, dashed trace). Instrument settings for the EPR spectroscopy: modulation amplitude, 0.79 millitesla. The g values are indicated in the figure.
up
Sequence motif-specific assignment of the Fe/S I and II centers in mammalian XOR
This study has established the unequivocal sequence motif-specific assignment of the two [2Fe-2S] clusters in recombinant XOR. The conserved cysteines (including Cys-43 and Cys-51) in the N-terminal plant-type ferredoxin motif in the Fe/S domain (Fig. 6, bottom, pink ribbon, and Fig. 7) serve as ligands to the Fe/S II center with unusual EPR properties, while the other conserved cysteines (including Cys-115) in the C-terminal unusual [2Fe-2S] protein fold motif (Fig. 6, bottom, blue ribbon, and Fig. 7) serve as ligands to the Fe/S I center with typical EPR properties of plant-type ferredoxins. This assignment is unexpected given the unusual EPR signal of the Fe/S II center and the unusual protein folding of the C-terminal subdomain (Fig. 6, bottom, blue ribbon) in XOR and the related enzymes. It is unequivocally established that the unusual EPR features of the Fe/S II center in dimeric XOR are not due to a polypeptide fold or a cluster-binding sequence motif. The faster spin-lattice relaxation time of the Fe/S II center for a regular plant ferredoxin-type [2Fe-2S]1+ cluster and the bovine XOR and D. gigas AOR structures imply a possible relation to the solvent accessibility of the N-terminal plant ferredoxin subdomain (Fig. 6, bottom, pink ribbon, and Fig. 7) and/or a magnetic interaction between the Fe/S I and Fe/S II centers (Figs. 6, 8).
It has been reported that the EPR signal attributed to the Fe/S I center of XOR and AOR interacts magnetically with the Mo center when the two sites are paramagnetic. More recently, using the local spin model, More et al. have assigned the reducible sites of the two [2Fe-2S]2+,+ clusters in D. gigas AOR, with the same conclusion that we reached here for the recombinant XOR by site-directed mutagenesis studies. The EPR spectroscopic heterogeneity of the Fe/S I center in demolybdo XOR dimer (Fig. 8) may be attributed to the absence of the Mo center in the recombinant dimeric enzymes used in this work.
Finally, after completion of this study, the crystal structure of bovine milk XDH was refined to 2.1 Å resolution. In conjunction with this structural information, the present sequence motif-specific assignment of the two [2Fe-2S] clusters in XOR indicates that the Fe/S I center is located in the vicinity of the Mo-pterin center and the Fe/S II center in the vicinity of the FAD center (Figs. 6, 7). Hence, the sequence of the intramolecular electron transfer in XOR probably occurs as follows: Mo-pterin → Fe/S I → Fe/S II → FAD (Fig. 6). These features appear to be common among several other XOR-related enzymes with similar cofactor arrangement and EPR properties as mammalian XOR.
*The recombinant XOR samples used in these EPR experiments (Figs. 8-13) have been prepared and characterized by Professor Takeshi Nishino and coworkers (Nippon Medical School). We thank Drs. K. Tamura, S. Oogami, and T. Iizuka (The Institute of Physical and Chemical Research (RIKEN), Wako-shi, Japan) for allowing us to utilize the EPR facility.
*One can follow the link to read the original article of this work.
 Go to the ICC Project Details Page
Go to the ICC Project Details Page